一組遺傳性凝血因數缺乏癥,由單個凝血因數缺乏所致的出血性疾病,較少見。在兒童期即有過度出血的表現:嚴重鼻衄、肌肉或關節出血或大瘀血斑。疾病的嚴重程度與所缺乏的凝血因數血漿水準成正比。血友病是一種X染色體伴性隱性遺傳性疾病,有陽性傢族史且隻影響男性發病。在遺傳性凝血因數缺乏癥中,以血管性假血友病最多見,其次為血友病。需經實驗室檢查才能確診。最主要的篩選試驗是凝血酶原時間(PT)和活化或白陶土的部分凝血活酶時間(APTT或KPTT)。在凝血因數缺乏性疾病中出血血時間一般正常,但血管性假血友病(VWD)有出血時間延長,因為血管性假血友病缺乏血管性假血友病因子(VWF),影響血小板功能,使初期止血受損(見血管性假血友病)。凝血酶原時間(PT)測定因子Ⅱ、Ⅶ、Ⅸ、Ⅹ活性,部分凝血活酶時間(APTT或KPTT)測定因子Ⅻ、Ⅺ、Ⅸ、Ⅷ和Ⅹ。延長的PT、APTT被正常血漿或血清糾正的模式可反映凝血因子缺乏的種類,若不能糾正則反映抗凝物質存在。
血友病甲和血友病乙 血友病甲即傳統所稱的血友病,是由於因子Ⅷ(FⅧ:C)缺乏致病。血友病乙(又名克裡斯馬斯氏病,PTC缺乏癥)是由於FⅨ缺乏致病。血友病甲的發病率是乙的7倍。發病率僅次於血管性假血友病。臨床上鑒別較難,需依靠實驗室檢查,治療須早期,足夠地補充所缺乏的凝血因子。
遺傳方式和病因 血友病甲、乙是性聯隱性遺傳性疾病,其遺傳基因位於X染色體上,男性發病,女性傳遞。血友病甲的FⅧ基因,現已提純並已知其序列。它是一種大基因,占X染色體長度的0.1%。70%的血友病甲有阻性傢族史,30%的病例是由於基因突變。血友病乙有明顯傢族史者少。故此基因似有高度的自發性突變率,使女性X染色體的一條隨機地無作用,不活化。患者與正常女子結婚其子正常,其女兒100%是血友病甲或乙傳遞者。傳遞者女子與正常男子結婚,其子半數為血友病患者,其女半數為傳遞者(見圖)。攜帶者(母親)的FⅧ:C血漿水平一般隻及正常婦女的50%(平均值為25~75%)。90%的血友病甲血漿不能中和天然的人抗因子Ⅷ抗體,稱為血友病甲交叉反應物質陰性 (CRM-)型即缺乏FⅧ:C,少數病人的血漿(CRM+)型,這反映患者的 FⅧ活性缺乏但有抗原性。現已知因子Ⅷ是一種糖蛋白,存在於科恩氏組分Ⅰ及冷沉淀物中,由二部分組成,分子量低的部分主要是因子Ⅷ的凝血活性(FⅧ:C)所在部位,缺乏時導致血友病甲,其抗原性(FⅧ:CAg)隻占FⅧ抗原性極小一部分;分子量高的部分為血管性假血友病因子(VWF,過去名為ⅧR:Ag)缺乏時導致血管性假血友病 (VWD)。FⅧ的抗原性大部分來自此大分子量部分。近年來的研究表明ⅧC是通過染色體X的基因遺傳。而 VWF則是通過常染色體遺傳,說明為何血友病甲的Ⅷ:C減少而Ⅷ:VWF正常。過去報道FⅧ:C減少,伴出血時間延長,目前認為就是VWD的變異型。文獻上尚有少數報道FⅧ:C缺乏伴FⅤ缺乏,或伴蛋白 C缺乏的病例,屬常染色體隱性遺傳,男女都可發病。血友病乙的攜帶者(母親)的因子Ⅸ水平平均隻有正常人的33%,約有10%的攜帶者低於正常的25%,因此血友病乙攜帶者有出血癥狀者較多。血友病乙交叉反應物質陽性(CRH+)型患者血漿中除缺乏因子Ⅸ活性外,其抗原性(交叉反應物質)也缺乏,不能中和抗因子Ⅸ抗體,若抗原性正常則稱為CRM+型,抗原性減少者稱為CRMR型血友病乙,變異型較多。

臨床表現 每一累及的傢族的臨床表現不同,但同一傢族中的患者缺乏的因子水平基本相似。血友病乙的臨床表現與血友病甲相同。輕型較多見,有的病人直到小手術或拔牙後出血不止,作進一步檢查時才被發現。血友病甲血漿 FⅧ:C活性測不到的患者常有嚴重的“自發”出血,本病的出血大多為創傷後出血,隻是創傷極輕微而未被註意,因而似乎是“自發”出血。出血部位常在較深的組織,包括關節、肌肉、腦、腹膜後血腫,出血深部腫脹及疼痛是主要的癥狀。血腫可引起組織壞死、外周神經病變、福爾克曼氏缺血性攣縮、關節畸形等癥狀。嚴重的血友病患者均在嬰兒或兒童期獲得診斷,若在中年後才首次發病的嚴重血友病,缺乏傢族史,則應進一步測定獲得性 FⅧ抗體、並除外血管性假血友病。曾有報道漿細胞或淋巴細胞性疾病,免疫性疾病可伴發FⅧ抗體。如類風濕性關節炎、聖路易腦炎(SLE)、潰瘍性結腸炎、單克隆γ球蛋白病和產後狀態、皰疹樣皮炎等。偶有一些報道抗因子Ⅷ抗體發生在無基礎疾病或藥物史的患者。輕度血友病患者,血漿有5~50%因子活性,可以正常生活,至嚴重外傷後才有出血表現,篩選試驗可以正常或正常低限。中度血友病者,血漿有2~5%Ⅷ或Ⅸ因子活性,常有自發性出血和小損傷後的過度出血,若發生嚴重外傷雖不一定合並深部出血,亦應該及時就醫,嚴重患者,血漿因子活性<1%,常因有明顯的出血癥狀或傢族史,故出生時即得出診斷。患者有終生的自發性出血(是輕度外傷後的出血),深部肌肉註射應屬禁忌。患者仍可接受預防性免疫接種,但要求接種時操作溫和,在局部註射處壓迫5分鐘,並在隨後數天內觀察註射部位有否出血癥象。患者因出血而需替代性治療時,亦應遵循上述操作要求。
診斷 男性患者反復關節出血或深部血腫形成,血漿FⅧ:C或FⅨ:C少於40%,有出血的性聯傢族史即可診斷血友病(甲或乙)。若FⅧ:C水平降低但傢族史不典型(或僅有2~3例男性出血患者)則血管性假血友病尚不能除外。
實驗室特點:血友病患者血管壁和血小板功能正常,因此出血時間(BT)正常。患者有正常的纖維蛋白原和因子Ⅱ、Ⅶ、Ⅴ活性,因此凝血酶原時間(PT)正常。但是有功能的凝血因子Ⅷ活性或Ⅸ缺乏,使內源性凝血系的試驗異常。診斷必須基於體外凝血活性的篩選試驗,部分凝血活酶時間(APTT或KPTT)或凝血活酶生成時間(比格斯氏TGT)。若患者BT、PT、TT(凝血酶時間)都正常而APTT延長,則有必要進一步測定血漿FⅧ:C或FⅨ水平,有助於按嚴重程度分型。FⅧ:C存在於正常及硫酸鋇或氫氧化鋁吸附的新鮮血漿中,但不存在於血清;FⅨ存在於正常血清而不存在於吸附血漿中,因此若APTT(KPTT)或比格斯氏 TGT可被正常吸附血漿糾正而不被正常血清糾正,則可定性診斷為血友病甲;若異常比格斯氏TGT可被正常血清糾正而不被吸附血漿糾正,則可定性診斷為血友病乙。血管性假血友病患者臨床癥狀相似於血友病,但兩性均可患病。患者血漿中缺乏馮·維勒佈蘭德氏因子(VWF),一種可結合於血小板膜並參與血小板粘附和血小板與血小板間相互反應的蛋白。VWF並且是FⅧ:C的攜帶蛋白,在血漿中與Ⅷ:C以非共價鍵結合而使FⅧ:C得到穩定。因此VWD患者由於VWF(Ⅷ:C攜帶分子)的減少或缺乏而使FⅧ:C半壽期縮短,患者FⅧ/VWF水平下降,同時伴FⅧ:C水平下降,出血時間延長,血小板對瑞絲托黴素不起聚集反應,因此與血友病不同(見表)。
血友病甲、血友病乙和血管性假血友病(VWD)的鑒別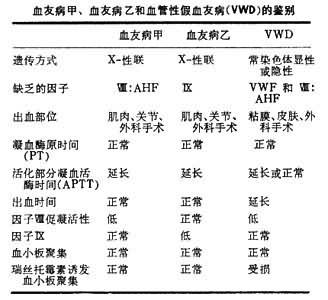
目前已對血友病攜帶者,在妊娠中期用一種特殊的小孔胎兒鏡來獲取純的胎兒血(不混有羊水或母血),作FⅧ:C和FⅧ:CAg測定可判斷胎兒是否為血友病患者。血友病患者FⅧC:Ag常與FⅧ:C活性呈一致性減少,少數可以正常,因此若胎兒血FⅧC:Ag值顯著低於10%可以診斷血友病甲,而正常則可以排除中至重度的血友病。近年來應用基因診斷方法,危險性小,正確率更高。隻是技術要求高,目前未得推廣。
治療 新鮮全血、血漿或新鮮冰凍血漿(FFP)可用於凝血因子的替代法治療。但需輸註的量大,且有導致高血容量的危險,甚至在大量輸註後血漿凝血因子仍不能達到足夠水平。此外,輸入的大量紅細胞和晶體將導致原有的凝血因子稀釋而加重出血傾向。因此在第一個血容量(5L)輸註時就應開始用濃縮因子制劑作替代性治療,後者以較小的容量而達到足夠的血漿水平。替代治療的劑量選擇,取決於血友病類型、出血的嚴重程度和所希望達到的血漿因子水平 (%)。因正常血漿中FⅧ或FⅨ平均水平為100%,起止血作用的最小有效水平在血友病甲為25~30%,血友病乙為20~25%,一般需將血漿因子水平提高到30%以上。
替代性治療中所需輸註的因子(凝血單位),可按每增高0.5%因子水平需輸註1單位Ⅷ:C/kg計算。如一男性患者體重60kg,希望達到的因子水平為50%,患者的基礎Ⅷ:C水平為10%,即需增高40%,則 所需因子(凝血單位)=60×40÷2=1200單位Ⅷ:C1凝血單位相當於1ml含有100%因子Ⅷ或Ⅸ活性的正常新鮮血漿。
因子Ⅷ半衰期8~12小時,輸入後大部分留在血管內,常需每日輸註2次。因子Ⅸ僅半數留在血管內,因此需要輸註雙倍量來達到同樣水平。FⅨ半衰期18~24小時,每日隻需輸註1次。
DDAVP治療:某些藥物如兒茶酚胺、胰島素等,可使正常人FⅧ:C和FⅧ:VWF升高3倍。DDAVP是一種合成的抗利尿激素,有上述相似的作用,但副作用更小。DDAVP用以治療輕中度血友病,使Ⅷ:C活性增高3~6倍。