一組少見的遺傳性糖原代謝紊亂的疾病。又稱糖原沉著癥、糖原累積病。因糖原分解與合成有關的某些酶系統缺乏,糖原分解困難,糖原異常地沉積於全身各組織,尤其是肝臟、心臟及肌肉中。表現為肝臟腫大,或伴有低血糖和高血脂、血清乳酸增高、心臟擴大、肌張力降低、腎腫大、高尿酸血癥及肌紅蛋白尿,中樞神經癥狀包括運動障礙、智力差。確診需依靠酶的測定。本病為常染色體隱性遺傳。常見於男性,多在嬰兒期發病,兒童期死亡,少數患者可活到成年。目前尚缺乏有效治療,主要是對癥處理。
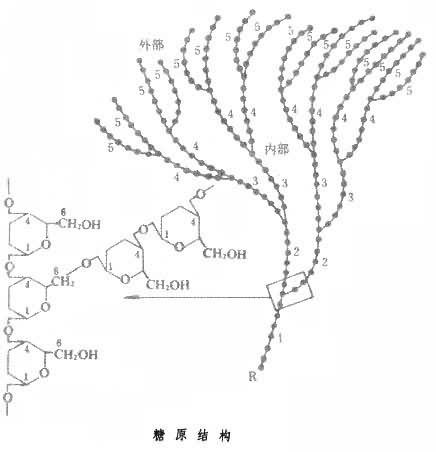
糖原代謝 人體內的碳水化合物以糖原形式儲存。糖原是分子量高達300~1000萬、由1萬個以上葡萄糖單位聚合而成的高分子多糖,有許多分支,而呈樹枝狀(見圖),直鏈部分以 α-1,4-糖苷鍵相連,側鏈處又以 α-1,6-糖苷鍵連接。糖原分子中心有葡萄糖殘基還原端,在分枝外周有許多非還原端,糖原的合成和分解就在非還原端進行。糖原分解需要磷酸化酶催化,最先是非還原端α-1,4-糖苷鍵的糖苷基端磷酸化,水解為1-磷酸葡萄糖,1-磷酸葡萄糖在磷酸葡萄糖變位酶催化下變為6-磷酸葡萄糖(G-6-P),然後G-6-P在6-磷酸葡萄糖酶影響下變為葡萄糖。糖原分解的另一途徑由細胞溶酶體中的 α-1,4-糖苷酶來完成。糖原生成從葡萄糖開始,首先葡萄糖由己糖激酶催化為6-磷酸葡萄糖,再經過磷酸葡萄糖變位酶催化,生成1-磷酸葡萄糖,1-磷酸葡萄糖在二磷酸尿嘧啶核苷 (UDP)、葡萄糖焦磷酸化酶影響下生成UDP-葡萄糖,然後經過糖原合成酶及分支酶協同催化下,最後形成糖原。組織(肝、骨骼肌、心肌、平滑肌)中的糖原等細胞內外液的葡萄糖經常保持動態平衡,保證葡萄糖不間斷地適當供應全身各組織器官,特別對腦組織是一刻不能缺少的。
臨床分型 依據缺陷的酶不同可分為10型及酶正常的Ⅺ型,又可根據糖原貯積的主要器官分為肝型、心型、肌型。其中以肝型較多見,肝型包括Ⅰ型、Ⅲ型、Ⅳ型、Ⅵ型、Ⅷ型;心型為Ⅱ型;肌型為Ⅴ型及Ⅶ型。癥狀的輕重取決於糖原轉化為血中葡萄糖的障礙程度。疾病的最後確診依靠酶測定。Ⅱ型、Ⅲ型、Ⅳ型可通過羊水穿刺測酶及羊水細胞培養作產前診斷。
①糖原貯積病Ⅰ型,又稱肝型糖原貯積病。1929年首先由E.O.K.馮·吉爾克描述,故又名馮·吉爾克氏病。常累及肝、腎。1952年發現本病患者肝內缺乏6-磷酸葡萄糖酶,造成糖原分解障礙,肝臟不能從糖原、乳酸、氨基酸形成葡萄糖,引起空腹血糖低,而低血糖抑制胰島素分泌,脂蛋白分解減少,脂蛋白酶活性降低,使脂肪大量動員,刺激脂肪分解,形成高甘油三酯血癥。由於糖酵解增多,形成乳酸增多。脂肪酸在肝內氧化不全,血中酮體增多,可出現酮癥酸中毒。由於慢性酸中毒引起負鈣平衡,可發生骨質疏松。又因己糖旁路代謝增強,尿酸合成增加,引起高尿酸血癥及痛風。本型主要侵犯嬰兒、兒童、成人,癥狀隨年齡有所不同,兩性均可受累。
診斷除根據臨床表現外,可作胰島高血糖素耐量試驗,肌註腎上腺素後血糖不升高,而乳酸增高,這有助於診斷。確診需作肝組織活檢,可見肝細胞腫大,糖原增加常超過5%,6-磷酸葡萄糖酶活性降低或消失。
飲食宜少量多餐飲,以避免低血糖和防止酸中毒。膳食中蛋白質含量正常,脂肪要低,總熱量不宜過高,以防止肥胖。血清乳酸高的病人宜服碳酸氫鈉,以防止酸中毒。有的病人可作門腔靜脈吻合術,以將血糖轉移至全身,可較好利用,這對糾正代謝紊亂與生長發育也有裨益。
②糖原貯積病Ⅱ型,又稱蓬普氏病。是最嚴重的一型。1963年發現。病因為溶酶體中 α-1,4-葡萄糖苷酶(即酸性麥芽糖酶)缺陷。α-1,4-葡萄糖苷酶存在於各組織細胞溶酶體內,缺乏此酶即不能分解糖原,以致糖原貯積在溶酶體內,實際上是容酶體貯積病。糖原大量累積於心肌、骨骼肌等全身組織,引起心臟增大,心臟重量可達正常的2~5倍,故又稱心型糖原累積病。患兒出生後半年之內出現紫紺、呼吸困難,嗆咳常見,常因心力衰竭或支氣管肺炎於1歲內死亡。成年患者癥狀輕微,表現為慢性全身肌無力,肌張力低下。診斷有賴於肝臟和肌肉活檢,電子顯微鏡檢查顯示糖原顆粒沉積增多,肝糖原顆粒外有一容酶體膜存在。肝、肌肉及白細胞內α-1,4-葡萄糖苷酶均缺乏,皮膚活檢後培養的纖維細胞中亦缺乏此酶,據此可確診。目前尚缺乏有效治療,有人研究用白細胞提取酶替代治療。
③糖原貯積病Ⅲ型,又稱科裡氏病。由於淀粉1,6-糖苷酶即脫支酶缺乏,而1-磷酸葡萄糖酶和磷酸化酶均正常,因此糖原分子隻能分解到第一層分支為止,不能完全分解,結果在許多組織中聚集結構異常的糖原。過多的糖原累積於肝、心肌和其他肌肉組織。本型比I 型癥狀要輕,但較常見。多見於幼兒,表現肝臟腫大,發育障礙,至青春期肝臟可恢復正常大小。血脂升高,但尿酸正常。成人癥狀較輕,僅有肌肉無力或無癥狀。由於糖原異生正常,故空腹低血糖少見。根據白細胞、紅細胞、肝臟、肌肉及心肌中淀粉1,6-糖苷酶缺乏即可確診。腎上腺素及胰高血糖素試驗接近正常。本型預後良好,主要對癥治療。
④糖原貯積病Ⅳ型,又稱安德森氏病。罕見,系淀粉1,4→1,6-轉糖酶即分支酶缺乏,所貯積的糖原結構異常,分支減少。臨床表現為進行性肝硬變、腹水、空腹血糖低、肝功能異常。病兒2歲左右夭折。若新生兒有肝硬變即應疑及本病。預後差,無特效治療。
⑤糖原貯積病Ⅴ型,又稱肌型糖原貯積病、麥卡德爾氏病。因肌磷酸化酶缺乏,肌糖原分解困難,糖原貯積,有關糖代謝的其他酶系統正常。患者以男性占多數,傢族中兄弟姐妹可患同樣疾病,患者往往有近親結婚史。一般兒童期無癥狀,待至10餘歲發病。由於酶缺乏,正常合成的糖原不能在肌肉內作為燃料,因此劇烈運動後出現肌肉酸痛、四肢僵硬、肌肉痙攣,有時引起肌紅蛋白尿,甚至腎功能衰竭。可作運動前後缺血性乳酸試驗,即以血壓繃帶維持血壓於收縮壓,同時使手指伸張握拳,比較運動前後血乳酸濃度,正常人運動後血乳酸濃度增高,而本病患者不升高,原因為缺乏磷酸化酶,肌糖原未能分解。肌肉活檢發現有肌糖原累積及肌磷酸化酶缺乏即可確診。無特效治療,平時避免劇烈運動,可防止發作。
⑥糖原貯積病Ⅵ型,又稱赫爾氏病。因肝缺乏磷酸化酶,不能分解肝糖原產生葡萄糖,維持血糖水平,並使肝糖原累積於肝。肌磷酸化酶正常。臨床表現以肝腫大為特征,低血糖癥狀一般較輕或缺如。診斷除依靠肝組織活檢及肝磷酸化酶分析外,白細胞磷酸化酶鑒定也有助於診斷。本型病情輕,預後好,可不予治療。
⑦糖原貯積病Ⅶ型,又稱垂井氏病。由於缺乏肌肉磷酸果糖激酶,糖原合成障礙,肝糖原太少。肌肉磷酸果糖激酶活性為正常的1~3%。癥狀似V型。重體力勞動後發生肌痙攣和肌疼痛。由於約50%紅細胞中的酶活性消失,紅細胞壽命縮短,有網織紅細胞增高。確診主要靠肌肉活組織檢查或查血紅細胞,鑒定此酶缺乏與否。本型預後較好,但無特效治療。
⑧糖原貯積病Ⅷ型,肝磷酸化酶激酶缺乏所致。為性聯遺傳,也有學者認為因肝磷酸化酶激酶呈失活狀態。磷酸化酶激酶可催化磷酸化酶b(無活性),使之轉變成磷酸化酶 a(活性型)。正常肝內60%磷酸化酶有活性,缺乏此酶,磷酸化酶b轉化為磷酸化酶a的數量降低,糖原分解發生障礙,肝糖原累積於肝和肌肉。表現無癥狀的肝腫大、肢體僵硬、去大腦狀態、肌無力、血清轉氨酶升高、血脂升高、低血糖,空腹血乳酸可正常,攝入碳水化合物後血乳酸升高,胰高血糖素試驗顯示正常反應。在白細胞、紅細胞及肝臟中缺乏磷酸化酶激酶b,即可確診。患者大多數為男性,一般至嬰幼兒期夭折。無特殊治療。
⑨糖原貯積病Ⅸ型。少見。肝磷酸化酶激酶缺陷,常累及肝臟,表現肝大,無低血糖酸中毒。為常染色體隱性遺傳。
⑩糖原貯積病 X型。少見。依賴cAMP的磷酸化酶激酶缺陷,常累及肝、肌肉,表現肝大,肝、肌肉糖原沉積。
⑪Ⅺ型。所有酶的活性均正常。常累及肝或腎,表現肝大,伴有范可尼氏綜合征、維生素D 抵抗性佝僂病、遺傳性果糖不耐受癥等。