一種X染色體伴性隱性遺傳性疾病。患者均為男性。
分類 分為血友病甲和血友病乙。前者即傳統所稱的血友病,是由於因數Ⅷ缺陷致病。後者是由於因數Ⅸ缺陷致病。前者的發病率是後者的7倍。
遺傳方式和病因 血友病甲和血友病乙均是性聯隱性遺傳性疾病。遺傳基基因位於X染色體上。由女性傳遞,男性發病。血友病甲的因子Ⅷ基因已提純並已知其序列。70%的血友病甲有陽性傢族史,30%的病例是由於基因突變。血友病乙有明顯傢族史者少,故此基因似有高度的自發性突變率(見圖)。
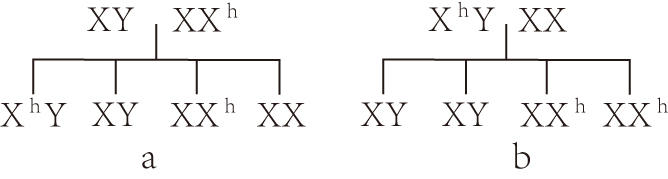 血友病的遺傳方式
血友病的遺傳方式
a 傳遞者女子與正常男子結婚,其子半數為血友病患者,其女半數為傳遞者 b 血友病患者與正常女子結婚,其子正常,其女兒100%是血友病傳遞者
臨床表現 每一累及傢族的臨床表現不同,但同一傢族中的患者缺乏的因子水平基本相似。臨床表現輕型者較多見,患者直到做小手術後出血不止,進一步檢查時才被發現。血漿因子Ⅷ:C活性測不到的血友病甲患者常有嚴重的出血。本病的出血大多為創傷後出血,出血部位常在較深的組織,包括關節、肌肉、腦、腹膜後血腫,血腫可引起組織壞死、外周神經病變、缺血性攣縮、關節畸形等癥狀。輕度血友病患者(血漿有5%~50%因子活性),至嚴重外傷後才有出血表現。中度血友病者(血漿有2%~5%Ⅷ或Ⅸ因子活性),常有自發性出血和小損傷後的過度出血,若發生嚴重外傷雖不一定合並深部出血,亦應該及時就醫。嚴重患者(血漿因子活性小於1%)常因有明顯的出血癥狀或傢族史,在出生時即得出診斷。
血友病甲、血友病乙和血管性假血友病(VWD)的鑒別| 血友病甲 | 血友病乙 | VWD | |
|---|---|---|---|
| 遺傳方式 | Ⅹ–性聯 | Ⅹ–性聯 | 常染色體顯性或隱性 |
| 缺乏的因子 | Ⅷ | Ⅸ | VWF和Ⅷ |
| 出血部位 | 肌肉、關節、手術 | 肌肉、關節、手術 | 黏膜、皮膚、手術 |
| 凝血酶原時間(PT) | 正常 | 正常 | 正常 |
| 凝血活酶時間(APTT) | 延長 | 延長 | 延長或正常 |
| 出血時間 | 正常 | 正常 | 延長 |
| 因子Ⅷ | 低 | 正常 | 低 |
| 因子Ⅸ | 正常 | 低 | 正常 |
| 血小板聚集 | 正常 | 正常 | 正常 |
| 瑞斯托黴素誘發的血小板聚集 | 正常 | 正常 | 受損 |
實驗室檢查 出血時間及凝血酶原時間正常。部分凝血活酶時間(APTT或KPTT)或凝血活酶生成時間(TGT)延長。進一步測定血漿因子Ⅷ或因子Ⅸ水平可確診及按嚴重程度分型。
診斷 男性患者,反復關節出血或深部血腫形成,血漿因子Ⅷ或因子Ⅸ少於40%,有出血的性聯傢族史即可診斷血友病。若因子Ⅷ水平降低,但傢族史不典型,則血管性假血友病尚不能除外。血管性假血友病患者血漿中缺乏馮·維勒佈蘭德氏因子(VWF),患者Ⅷ/VWF水平下降,同時伴FⅧ水平下降,出血時間延長,血小板對瑞斯托黴素不起聚集反應,因此與血友病不同(見表)。
治療 新鮮血漿或新鮮冰凍血漿(FFP)可用於凝血因子的替代法治療。但需輸註的量大,甚至在大量輸註後血漿凝血因子仍不能達到足夠水平。采用因子濃縮劑(如因子Ⅷ濃縮劑和凝血酶原復合物)行替代治療,可用相對小的容量達到理想的血漿凝血因子水平。替代治療的劑量選擇,取決於血友病類型、出血的嚴重程度和所希望達到的血漿因子水平(30%以上)。因患者有自發性出血,深部肌肉註射應屬禁忌,采用替代性治療時要求操作溫和,註射後在局部壓迫5分鐘,並在隨後數天內觀察註射部位有否出血征象。