苯丙氨酸羥化酶基因缺陷引起的遺傳性氨基酸代謝障礙性疾病。常染色體隱性遺傳。最早報告於1934年。臨床特徵是嚴重的智力發育障礙(白癡)及尿中有過量苯丙酮酸排出。對多數病例出生後單純控制飲食中苯丙氨酸的攝入,即可成功地防止腦損害。新生兒患者可無癥狀,但若延誤治療一年,即可發生不可逆的腦損害,故早期診斷和治療極為重要。發病率在世界各地差異很大,黑種人中罕見,在中國發病率為1:16000。
發病機機理 苯丙氨酸是必需氨基酸之一。人體攝入的苯丙氨酸除用於蛋白質合成外,在肝臟苯丙氨酸羥化酶及其輔因子的作用下生成酪氨酸,少量苯丙氨酸進入次要代謝途徑,經轉氨基作用變成苯丙酮酸。酪氨酸也是人體必需的氨基酸,可用以合成多巴胺、腎上腺素、甲狀腺素等重要激素和神經遞質及黑素,也可進一步代謝生成尿黑酸進入三羧酸循環為機體提供能量。有些人因遺傳基因缺陷,其苯丙氨酸羥化酶(或輔因子)缺乏或活性減低,苯丙氨酸主要代謝途徑受阻,血液中遊離苯丙氨酸濃度異常升高,次要代謝途徑活躍,致正常代謝時數量很少的產物苯丙酮酸在體液中明顯增多,於是出現高苯丙氨酸血癥及苯丙酮酸尿癥(見圖)。據信引起新生兒中樞神經系統分化停滯或發育遲緩的有害物質可能就是苯丙氨酸本身。發病機理是升高的苯丙氨酸與其他氨基酸在進入神經細胞時競爭轉運系統,致神經細胞內氨基酸不平衡,從而抑制蛋白質合成及神經突觸的形成,影響微管(紡錘絲的基本成分)的形成,造成大腦的不可逆損害。
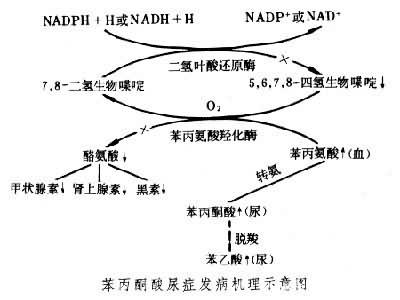
苯丙氨酸可通過代謝旁路生成苯乳酸→苯乙酸→苯乙酰谷氨酰胺,這些代謝產物經汗及尿排出,使汗、尿帶鼠尿或馬糞氣味。
分型及臨床表現 苯丙酮酸尿癥患者的血液中苯丙氨酸增高。高苯丙氨酸血癥分5型。經典PKU是Ⅰ型,此型病人出生時很少有癥狀,生後第1年的早期患兒身上可有類似鼠尿的黴臭味。苯丙氨酸的正常代謝產物酪氨酸減少,黑素合成缺乏必要的原料,故患兒毛發、虹膜及皮膚色素很淡。以上癥狀多不為雙親註意。1年以後出現明顯的智力障礙、癡呆、不會走路及說話、多動、癲癇樣發作、易激惹或有攻擊行為。此外,還可出現多汗、流涎、震顫、小頭畸型、牙釉質發育不良、生長速度減慢等。此型病人苯丙氨酸羥化酶幾乎全無活性。Ⅱ、Ⅲ型病人尿中無苯丙酮酸,輕者無癥狀而重者似Ⅰ型。此二型的特點是苯丙氨酸食物耐量正常,並以此區別於典型的PKU。Ⅳ、Ⅴ型病例出生不久,即有吞咽困難,因而很難喂養,半年內生長明顯遲鈍、肌肉痙攣、多涎,1年內發生癲癇。其病因為苯丙氨酸羥化酶的天然輔因子再生系統有缺陷,二氫蝶啶還原酶缺乏,不僅苯丙氨酸羥化障礙,腦內5-羥色胺及左旋多巴形成也受嚴重影響,故神經系統合並癥嚴重。
診斷 出現典型癥狀後診斷不難,但腦損傷後作出診斷為時已晚,為及時作出診斷,唯一可靠的方法為在新生兒中篩選高苯丙氨酸血癥病例。篩選成功的關鍵是:①測定全部新生兒。②在嬰兒能攝入蛋白質(哺乳)後進行。因 PKU嬰兒出生時血中苯丙氨酸可在正常水平。檢查方法為格思裡氏試驗(血中含苯丙氨酸時,枯草桿菌的生長即不能為β-2-噻吩丙氨酸所抑制)。PKU與其他類型高苯丙氨酸血癥的鑒別靠苯丙氨酸食物耐量。輔因子缺陷型病例的診斷需更特異的方法,包括酶學測定或補充相應輔因子後測血中苯丙氨酸水平。基因診斷技術的進步使PKU胎兒產前基因診斷成為可能。
治療 唯一切實可行的治療方法是限制飲食中苯丙氨酸的攝入。市售低苯丙氨酸半合成食品可將每日苯丙氨酸攝入量限制在250~500mg之內。嬰兒出生後60天內給予治療者,體力和智力發育可正常。療程要足夠長,現傾向於8~10歲後可慢慢放松嚴格控制,但若病情反復,則應重新恢復治療。有輔因子缺陷的病例治療困難,補充四氫生物蝶啶很難實現,因為它不能有效地穿過細胞或腦組織,單純低苯丙氨酸飲食不能控制癥狀。PKU的女性患者妊娠期應繼續治療,否則可致嬰兒小頭畸型及智力遲鈍。因苯丙氨酸降解主要在肝臟進行,故有人建議用肝移植的方法為患者提供穩定的酶源來治療本病,這在將來也許可行。